Alignments
How to align sequences and managing alignments.
 |
The alignment of nucleotide, RNA or amino acid sequences is one of the most common tasks in Bioinformatics.
The motivation for doing alignments is that high sequence similarity usually implies significant functional
or structural similarity. The simultaneous alignment of three or more sequences is an essential precondition
to phylogenetic analyses or studies about protein families. SeqAn provides two kinds of alignment data structures,
a matrix-like alignment data structure and a graph-based alignment representation.
Although there is a multitude of different alignment algorithms the basic syntax for performing an
alignment is always the same:
TScore result = localAlignment(TSequences, TScore, TAlgorithm)
TSequences, TScore, and TAlgorithm are just placeholders explained below. An actual call could look like this:
In this case the sequences are stored in alignment_ds . The chosen Gotoh algorithm uses affine
gap costs and it is configured with a scoring scheme, where matches are scored +1, mismatches -1, a gap-opening -2,
and a gap-extension -1. The globalAlignment call returns the score of the alignment and stores the actual
alignment in alignment_ds , which could be an Alignment Graph or an Align data structure.
If it is an alignment graph, its textual representation in PipMaker format
is illustrated in the following figure.
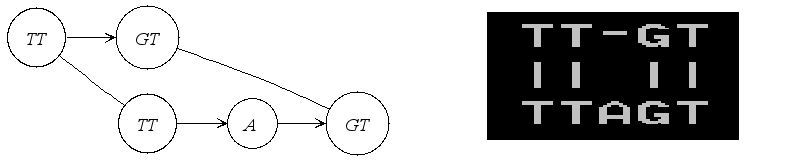 |
If one is only interested in the score of an alignment, a StringSet that holds the sequences is sufficient.
For instance, in the above example
would do the job. To sum it up, TSequences must be either an Align or an Alignment Graph containing the sequences, or a
simple StringSet. Note that there are also other possibilities, e.g., to retrieve just the matches of
an alignment or to print the traceback in the console.
TScore is a scoring object. The most common scoring objects are illustrated below in conjunction with different algorithms:
int score0 = globalAlignment(alignment_graph, score_type, NeedlemanWunsch() );
Blosum62 score_type_blossum(-1,-11);
int score1 = globalAlignment(alignment_graph, score_type_blossum, Gotoh() );
Score<int, Pam<> > pam(250, -1, 0);
int score2 = globalAlignment(alignment_graph, pam, Hirschberg() );
Note that certain algorithms require different score types, e.g., for an affine scoring scheme Gotoh and Hirschberg
work well but Needleman-Wunsch fails because it uses linear gap penalties.
By now you have seen the most common possibilities
for TScore and TAlgorithm . For local alignments TAlgorithm can be substituted with SmithWaterman() , which implements the well-known Smith-Waterman algorithm.
You can find more example code about computing global and local alignments here: Global Alignments, Local Alignments.
However, in some situations more sophisticated alignment algorithms are necessary. For example, in assembly projects one is usually not
interested in ordinary global alignments but so-called overlap alignments. These require a special initialization of the dynamic programming
matrix. Also semi-global alignments that try to fit one sequence into the other require adapted dynamic programming matrices.
That's why graph-based alignment algorithms can be configured with an AlignConfig object that has 4 boolean parameters.
The parameters indicate whether the first row and / or column is initialized with 0's and whether the maximum is searched
in the last row and / or column. So in total there are 2 to the power of 4 = 16 different configurations.
The following code snippet initializes the first row with 0's and looks for the maximum in the last row (classical
sequence fitting).
typedef StringSet<TString, Dependent<> > TStringSet;
typedef Graph<Alignment<TStringSet, void> > TGraph;
TStringSet str;
TString str0("TarfieldandGarfieldarestupid.");appendValue(str, str0);
TString str1("Garfield");appendValue(str, str1);
Score<int> score_type = Score<int>(2,-1,-1,-4);
TGraph g(str);
AlignConfig<true,false,false,true> ac;
int score = globalAlignment(g, score_type, ac, Gotoh() );
Multiple sequence alignments are part of SeqAn and currently under
development. If needed contact the SeqAn team.
SeqAn - Sequence Analysis Library - www.seqan.de