Class
ClusterReductionSpecialization for ReducedAminoAcid
Specialization for ReducedAminoAcid
| Defined in | seqan/reduced_aminoacid.h |
|---|---|
| Signature |
template <unsigned char n, unsigned char m = 24, typename TMatrix = Blosum62>
struct ClusterReduction;
|
Template Parameters
n |
the size of the reduced alphabet (between 2 and m-1) |
|---|---|
m |
size to truncate alphabet to, before clustering (one of 20, 22, 24; default 24) |
TMatrix |
Matrix used for clustering (default Blosum62, none other supported right now) |
Detailed Description
WhenToUse
Use m = 24 when you expect 'X' and '*' in the dataset you reduce from. This is especially the case on translated genomic reads.
If you have validated protein sequences, you can use can use m = 20 or m = 22, which will not include special characters (see AminoAcid for details).
Background
The method employed for reducing the alphabet is similar to Murphy et al, 2000, http://www.ncbi.nlm.nih.gov/pubmed/10775656
Correlation coefficients for the Blosum62 scores of all pairs of amino acids in the alphabet were computed and clustered with WPGMA (using UPGMA as second criterium when WPGMA yields the same distance between two clusters).
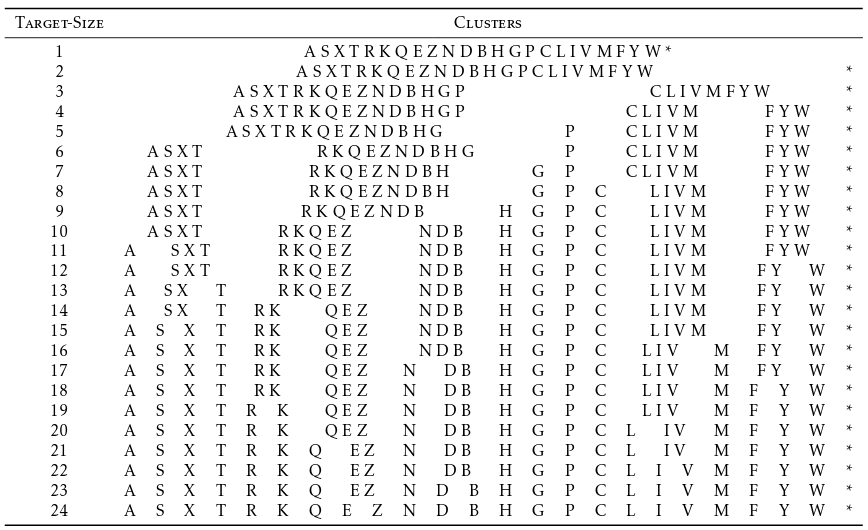
The exact clustering for m = 24.