|
SeqAn3 3.2.0
The Modern C++ library for sequence analysis.
|


|
|
SeqAn3 3.2.0
The Modern C++ library for sequence analysis.
|
|

Provides the algorithmic components for the computation of pairwise alignments. More...
 Collaboration diagram for Pairwise:
Collaboration diagram for Pairwise:Classes | |
| class | seqan3::alignment_result< alignment_result_value_t > |
| Stores the alignment results and gives access to score, alignment and the front and end positionss. More... | |
Functions | |
| template<typename sequence_t , typename alignment_config_t > requires detail::align_pairwise_single_input<sequence_t> && std::copy_constructible<std::remove_reference_t<sequence_t>> && detail::is_type_specialisation_of_v<alignment_config_t, configuration> | |
| constexpr auto | seqan3::align_pairwise (sequence_t &&seq, alignment_config_t const &config) |
| Computes the pairwise alignment for a pair of sequences or a range over sequence pairs. More... | |
Provides the algorithmic components for the computation of pairwise alignments.
Pairwise sequence alignments can be computed with the free function seqan3::align_pairwise. This function is called in the default case with a sequence pair and an alignment configuration object. Note the type of the pair must model seqan3::tuple_like, e.g. std::tuple or std::pair, with exactly two elements. The algorithm accesses the sequences via the corresponding get interface. Furthermore, the sequences stored in the pair must model std::ranges::viewable_range. This means the type returned by the get interface models either std::ranges::view or std::ranges::range and is an lvalue reference. If you don't know yet what a view or a range is, it is recommended to read through the ranges tutorial. The following code snippet demonstrates a simple use of the pairwise alignment interface.
In this snippet a global alignment over two nucleotide sequences is computed. Here the helper function std::tie is used to pass the two sequences as a tuple to the alignment algorithm. The special interface of std::tie allows to forward the two sequences as lvalue references such that no copy of the data is involved.
There are a lot of applications that need to compute many pairwise sequence alignments. Accordingly, the seqan3::align_pairwise interface offers an overload for ranges over sequence pairs. The following snippet shows a simple use case.
In SeqAn the alignment algorithm can be configured in many different ways. The core of this configuration are the different configuration elements that select specific features of the algorithm. To allow a maximal flexibility the configuration is separated from the alignment interface. This means the algorithm must be configured before it is invoked. The respective alignment configurations are defined in their own namespace called seqan3::align_cfg. This namespace is used to disambiguate configurations for the alignment algorithm with configurations from other algorithms in SeqAn.
To compute a pairwise alignment at least two configuration elements must be provided: The alignment method and the scoring scheme. Thus, a valid alignment configuration must specify what kind of alignment shall be computed, as it strongly depends on the corresponding context. A default wouldn't make much sense here. For similar reasons the scoring scheme has to be always provided by the user.
There have been numerous adaptions and modifications of the original global alignment problem to solve similar problems such as the local alignment. Here, the goal is to find a maximal homologue region between two sequences that has been conserved during the evolution. Other examples are the semi-global alignment which is frequently used in read mapping in order to align a smaller sequence into the context of a larger reference sequence.
The standard global and local alignments can be configured using seqan3::align_cfg::method_global and seqan3::align_cfg::method_local, respectively.
A semi-global alignment can be computed by specifying the free end-gaps in the constructor of seqan3::align_cfg::method_global. The parameters enable, respectively disable, the scoring of leading and trailing gaps at the respective sequence ends ( first sequence leading, second sequence leading or first sequence trailing, second sequence trailing gaps). The SeqAn alignment algorithm allows any free end-gap settings making it a very versatile algorithm. This option, however, is not available for the local alignment where penalising gaps at the ends of the sequences is always disabled.
Global Alignment:
Finding the optimal global alignment of two sequences is solved by the Needleman-Wunsch algorithm.
Semi-global Alignment (e.g. overlapping sequences):
The semi-global alignment is a specially configured global alignment, namely we do not penalize gaps at the ends of the alignment. Semi-global alignments are often used in genome assembly applications when trying to find matching overlaps.
Local Alignment (better suited to find conserved segments):
A local alignment is effectively a global alignment of two partial sequences. For example when two genes from different species are similar in short conserved regions and dissimilar in the remaining regions. A global alignment would not find the local matching because it would try to align the entire sequence. This is solved by the Smith-Waterman algorithm.
To compute an alignment a scoring and a gap scheme must be provided which give a "score" for substituting, inserting, or deleting a base within the alignment computation. Throughout SeqAn, a positive score implies higher similarity and/or a closer relatedness and a lower or even negative score implies distance. If you are used to dealing with "penalties" or "distances", instead think of "negative scores" when using SeqAn interfaces.
Scoring two letters of a single alphabet (or two similar alphabets) is performed by scoring schemes. A scoring scheme is any type that models seqan3::scoring_scheme_for, i.e. it must provide a member function that takes the two letters and returns the scheme-specific score. Algorithms that expect a scoring scheme should check this concept with their respective alphabet(s).
Two generic scoring schemes are available in SeqAn:
These also support scoring two nucleotides/amino acids of different types and they also support modification of their scores via set_() functions and by returning references to their internal score matrix. You can however add completely different types, as long as they model seqan3::scoring_scheme_for for the respective sequence alphabet type. In fact, when invoking the seqan3::align_pairwise interface it will be checked at compile time if the provided scoring scheme can be used in combination with the passed sequences and if not a static assertion is raised.
The scoring scheme can be configured with the seqan3::align_cfg::scoring_scheme element. Since the scoring scheme is strongly coupled on the sequences to be aligned, there is no default for it. Thus, it is mandatory for the developer to specify this configuration.
Throughout SeqAn we use the term gap to refer to an individual gap (see Gap) and a gap interval to refer to a stretch of consecutive gaps. When aligning two sequences a gap is introduced to mark an insertion or deletion with respect to the other sequence. However, because it is widely recognised that the likelihood of n consecutive gaps is much higher than that of n individual gaps the scoring of an individual gap or a stretch of gaps is not handled by the scoring scheme. This is based on the assumption that one biological event often introduces more than one gap at a time and that single character gaps are not common due to other biological factors like frame preservation in protein-coding sequences.
SeqAn offers the additional seqan3::gap_cost_affine which can be used to set the scores for opening (seqan3::align_cfg::open_score) or extending a gap (seqan3::align_cfg::extension_score).
Configurations can be combined using the |-operator. If a combination is invalid, a static assertion is raised during the compilation of the program. It will inform the user that some configurations cannot be combined together into one alignment configuration. In general, the same configuration element cannot occur more than once inside of a configuration specification. The following table shows which combinations are possible.
| Config | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0: Band | ❌ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ |
| 1: Gap scheme affine | ✅ | ❌ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ |
| 2: Min score | ✅ | ✅ | ❌ | ✅ | ❌ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ |
| 3: Method global | ✅ | ✅ | ✅ | ❌ | ❌ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ |
| 4: Method local | ✅ | ✅ | ❌ | ❌ | ❌ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ |
| 5: Alignment output | ✅ | ✅ | ✅ | ✅ | ✅ | ❌ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ |
| 6: End positions output | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ❌ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ |
| 7: Begin positions output | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ❌ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ |
| 8: Score output | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ❌ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ |
| 9: Sequence1 id output | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ❌ | ✅ | ✅ | ✅ | ✅ | ✅ |
| 10: Sequence2 id output | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ❌ | ✅ | ✅ | ✅ | ✅ |
| 11: Parallel | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ❌ | ✅ | ✅ | ✅ |
| 12: Score type | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ❌ | ✅ | ✅ |
| 13: Scoring scheme | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ❌ | ✅ |
| 14: Vectorised | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ✅ | ❌ |
The seqan3::align_pairwise interface returns a seqan3::algorithm_result_generator_range. This range is a lazy single pass input range over the computed alignments and the range's element types are seqan3::alignment_result objects. Even if only a single alignment is computed a range will be returned since it could be possible that one alignment invocation produces multiple results, e.g. to receive suboptimal alignments. The seqan3::alignment_result object contains only the information that has been requested via the output configuration (see below). The algorithm will then choose the most efficient implementation to compute the requested outputs. The following table shows the different output configurations:
| Output option | Available result |
|---|---|
| seqan3::align_cfg::output_score | alignment score |
| seqan3::align_cfg::output_end_position | end positions of the aligned sequences |
| seqan3::align_cfg::output_begin_position | begin positions of the aligned sequences |
| seqan3::align_cfg::output_alignment | alignment of the two sequences |
| seqan3::align_cfg::output_sequence1_id | id of the first sequence |
| seqan3::align_cfg::output_sequence2_id | id of the second sequence |
The begin and end positions refer to the begin and end positions of the slices of the original sequences that are aligned. For example, the positions reported for the global alignment correspond to the positions of the original sequences since the entire sequences are encompassed by the global alignment. In case of a local alignment the aligned part might only encompass a part of the original sequences. In this case, the begin and end positions denote the begin and end of the slices of the original sequences that are aligned. To obtain the actual alignment the option seqan3::align_cfg::output_alignment has to be specified. The options can be combiend with each other in order to customise the alignment algorithm and the respective output of the alignment. For example computing the alignment will always incur some run time penalty compared to just computing the score. See the following snippet for some examples:
If none of the above configuration was set by the user, then all output options will be enabled by default, i.e. the alignment algorithm will compute every output. Otherwise, if any of the output configurations was set by the user, then only the configured ones are available in the final seqan3::alignment_result. Trying to access an output which has not been configured will raise a static assertion informing the developer about the invalid access.
Since both algorithms (Smith-Waterman and Needleman-Wunsch algorithm) are based on dynamic programming, they run in quadratic time and memory O(nm) (where n and m are the lengths of the respective aligned sequences).
To reduce the time complexity you can use a banded alignment. It reduces the runtime by a constant although remaining quadratic and limiting the possible solutions slightly. You can speed up the computation significantly if you parallelize and simdify your alignment. More about banded and parallelization can be read below.
By default a generic alignment algorithm is used that supports all valid alignment configurations but for some special combinations of parameters a notably faster algorithm is available. It is automatically selected if all of the following requirements are satisfied:
There is a special shortcut for the above required scoring/gap configurations called seqan3::align_cfg::edit_scheme, which can be used to safe some typing.
The edit configuration can be further specialised with following configs:
SeqAn offers the computation of banded alignments to reduce the running time of the algorithm. This can be helpful if the region in which the optimal alignment exists is known a priori. To specify the banded alignment the developer can use the seqan3::align_cfg::band_fixed_size option.
This band configuration is initialised with a seqan3::align_cfg::lower_diagonal and a seqan3::align_cfg::upper_diagonal. The term diagonal is used to describe the position of the band boundary within the alignment matrix. The given value represents the offset that the lower, respectively upper, diagonal is shifted from the main diagonal, which starts in the origin of the alignment matrix. Accordingly, a negative value shifts the band boundary downwards in the alignment matrix and a positive value shifts the band boundary to the right.
The band parameters might be restricted depending on the configured alignment algorithm, e.g. the origin of the alignment matrix and the sink (the last cell in the last column) must be covered by the band when a global alignment is ought to be computed.
In general, the upper diagonal must always be greater than or equal to the lower diagonal to specify a valid band.
SeqAn's pairwise sequence alignment algorithm is internally accelerated using multi-threading. The parallel execution can be selected by specifying the seqan3::align_cfg::parallel configuration element. This will enable the asynchronous execution of the alignments in the backend. For the user interface nothing changes as the returned seqan3::algorithm_result_generator_range will preserve the order of the computed alignment results, i.e. the first result corresponds to the first alignment given by the input range. By default, a thread pool with std::thread::hardware_concurrency many threads will be created on a call to seqan3::align_pairwise and destructed when all alignments have been processed and the seqan3::algorithm_result_generator_range goes out of scope. The configuration element seqan3::align_cfg::parallel can be initialised with a custom thread count which determines the number of threads that will be spawned in the background.
Note that only independent alignment computations can be executed in parallel, i.e. you use this method when computing a batch of alignments rather than executing them separately.
Depending on your processor architecture you can gain a significant speed-up.
In some cases, for example when executing the alignments in parallel, it can be beneficial for the performance to use a continuation interface rather than collecting the results first through the seqan3::algorithm_result_generator_range. To be more precise, if more work needs to be done after the alignment has been computed, it could be better to stay within the thread and continue the work rather than buffering the result and computing the next alignment. The alignment algorithm allows the user to specify their own callback function which will be invoked by the alignment algorithm when a seqan3::alignment_result has been computed. To do so, the seqan3::align_cfg::on_result configuration element can be used during the alignment configuration. Note that if seqan3::align_cfg::on_result is specified, the algorithm seqan3::align_pairwise does not return a seqan3::algorithm_result_generator_range anymore. In fact, the algorithm's return type is void. The following code snippet illustrates this behavior:
|
no-apiconstexpr |
Computes the pairwise alignment for a pair of sequences or a range over sequence pairs.
| sequence_t | The type of sequence pairs (see details for more information on the type constraints). |
| alignment_config_t | The type of the alignment configuration; must be a seqan3::configuration. |
| [in] | seq | A tuple with two sequences or a range over such tuples. |
| [in] | config | The object storing the alignment configuration. |
This function computes the pairwise alignment for the given sequences. During the setup phase the most efficient implementation is selected depending on the configurations stored in the given seqan3::configuration object. The configuration also holds settings for parallel or vectorised execution.
In cases where only a single alignment is to be computed, the two sequences can be passed as a pair. The pair can be any class template that models the seqan3::tuple_like concept. The tuple elements must model std::ranges::viewable_range and std::copy_constructible. The following example demonstrates how an alignment is computed from a std::pair of sequences.
Alternatively, you can use std::tie as shown in the example below:
In many cases one needs to compute multiple pairwise alignments. Accordingly, the align_pairwise interface allows to pass a range over sequence pairs. The alignment algorithm will be configured only once for all submitted alignments and then computes the alignments sequentially or in parallel depending on the given configuration. Since there is always a certain amount of initial setup involving runtime checks required, it is advisable to pass many sequence-pairs to this algorithm instead of repeatedly calling it with a single pair.
For each sequence pair one or more seqan3::alignment_results can be computed. The seqan3::align_pairwise function returns an seqan3::algorithm_result_generator_range which can be used to iterate over the alignments. If the vectorised configurations are omitted the alignments are computed on-demand when iterating over the results. In case of a parallel execution all alignments are computed at once in parallel when calling begin on the associated seqan3::algorithm_result_generator_range.
The following snippets demonstrate the single element and the range based interface.
Strong exception guarantee.
Might throw std::bad_alloc if it fails to allocate the alignment matrix or seqan3::invalid_alignment_configuration if the configuration is invalid. Throws std::runtime_error if seqan3::align_cfg::parallel has been specified without a thread_count value.
The complexity depends on the configured algorithm. For the edit distance the following worst case over two input sequences of size 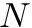 can be assumed:
can be assumed:
| Computing | Runtime | Space |
|---|---|---|
| score | 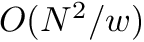 | 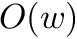 |
| back coordinate | | |
| front coordinate | | |
| alignment | | |
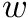 is the size of a machine word.
is the size of a machine word.
For all other algorithms that compute the standard dynamic programming algorithm the following worst case holds:
| Computing | Runtime | Space |
|---|---|---|
| score | 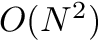 | 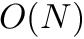 |
| back coordinate | | |
| front coordinate | | |
| alignment | | |
In the banded case the worst case is modified as follows:
| Computing | Runtime | Space |
|---|---|---|
| score | 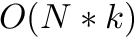 | 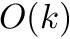 |
| back coordinate | | |
| front coordinate | | |
| alignment | | |
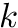 is the size of the band.
is the size of the band.
This function is re-entrant, i.e. it is always safe to call in parallel with different inputs. It is thread-safe, i.e. it is safe to call in parallel with the same input under the condition that the input sequences do not change when being iterated over.