|
SeqAn3
3.0.1
The Modern C++ library for sequence analysis.
|


|
|
SeqAn3
3.0.1
The Modern C++ library for sequence analysis.
|
|
Learning Objective:
In this tutorial you will learn how to compute pairwise sequence alignments with SeqAn. This tutorial is a walkthrough with links to the API documentation and is also meant as a source for copy-and-paste code.
| Difficulty | Intermediate |
|---|---|
| Duration | 60-90 min |
| Prerequisite tutorials | Quick Setup (using CMake) Alphabets in SeqAn Ranges |
| Recommended reading |
Aligning biological sequences is a very prominent component in many bioinformatics applications and pipelines. Well known genomic applications that use pairwise sequence alignments are read mapping, genome assembly, variant detection, multiple sequence alignment as well as protein search.
The goal of the pairwise alignment is to obtain an optimal transcript that describes how two DNA sequences are related to each other by means of substitutions, insertions, or deletions. The computed transcript describes then the operations necessary to translate the one sequence into the other, as can be seen in the following picture.

The alignment problem is solved with a dynamic programming (DP) algorithm which runs in 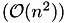 time and space. Besides the global alignment approach many more variations of this DP based algorithm have been developed over time. SeqAn unified all of these approaches into a single DP core implementation which can be extended easily and thus, with all possible configurations, is a very versatile and powerful tool to compute many desired alignment variants.
time and space. Besides the global alignment approach many more variations of this DP based algorithm have been developed over time. SeqAn unified all of these approaches into a single DP core implementation which can be extended easily and thus, with all possible configurations, is a very versatile and powerful tool to compute many desired alignment variants.
Let us first have look at an example of computing a global alignment in SeqAn.
In the above example we want to compute the similarity of two seqan3::dna4 sequences using a global alignment. For this we need to import the dna4 alphabet, the seqan3::nucleotide_scoring_scheme and the seqan3::align_pairwise in the beginning of our file. We also import the seqan3::debug_stream which allows us to print various types in a nice formatted manner.
In the beginning of the file we are defining our two sequences DNA sequences first_seq and second_seq. If you feel puzzled about what the _dna4 suffix does we recommend to read the Alphabets in SeqAn and Ranges before. In line 16-17 we configure the alignment job with the most simplistic configuration possible. In this case it is a global alignment with edit distance. Later in this tutorial we will give a more detailed description of the configuration and how it can be used. The minimum requirement for computing a pairwise alignment is to specify the seqan3::align_cfg::mode and the seqan3::align_cfg::scoring configuration elements. The first one selects the internal algorithm and the second one provides the scoring scheme that should be used to score a pair of sequence characters.
Now we are going to call seqan3::align_pairwise. This interface requires two arguments: a tuple or a range of tuples with exactly two elements and the configuration object. Independent of the number of pairwise alignment jobs submitted to the algorithm it always returns a range over seqan3::alignment_result. Later we will see how we can use a continuation interface which calls a user-defined function rather than iterating over the results sequentially. Finally, we output the score for the computed alignment.
pairwise_align does nothing as it returns a range which is evaluated in a lazy manner. Only when calling begin or incrementing the iterator over the range the alignment computation is invoked.
First we create the vector of seqan3::dna4 sequences. We keep the configuration as is and then modify the initial code to a range-based for loop looping over the alignment results. Since the seqan3::alignment_result is a class template and the template parameters are determined during the configuration step we use auto as the result type. The current result is cached inside of the lazy range and we capture the result as const & in order to not tamper with the result values.
Congratulations, you have computed your first pairwise alignment with SeqAn! As you can see, the interface is really simple, yet the configuration object makes it extremely flexible to conduct various different alignment calculations. In the following chapter you will learn more about the various configuration possibilities.
The configuration object is the core of the alignment interface. It allows to easily configure the alignment algorithm without changing the interface of the pairwise alignment. It uses a seqan3::configuration object to chain different configuration elements together using the logical or-operator ('|'-operator). You can find an overview over the available configurations here. The configurations for the alignment module are available in:
The configuration elements are all classes that wrap the actual information necessary for the configuration of the alignment algorithm. Depending on the configuration specification certain features of the algorithm are enabled or disabled. Moreover, during the initialisation of the algorithm the best implementation is chosen based on the given configurations. To avoid possible ambiguities with the configurations of other algorithms, the configuration elements for the alignment are defined in the special namespace seqan3::align_cfg.
The most straightforward algorithm is the global alignment which can be configured using the seqan3::align_cfg::mode with seqan3::global_alignment as the constructor argument.
The global alignment can be further refined by setting the seqan3::align_cfg::aligned_ends option. The seqan3::align_cfg::aligned_ends class specifies wether or not gaps at the end of the sequences are penalised. In SeqAn you can configure this behaviour for every end (front and back of the first sequence and second sequence) separately using the seqan3::end_gaps class. This class is constructed with up to 4 end gap specifiers (one for every end):
These classes can be constructed with either a constant boolean (std::true_type or std::false_type) or a regular bool argument. The former enables static configuration of the respective features in the alignment algorithm. The latter allows to configure these features at runtime. This makes setting these values from runtime dependent parameters, e.g. user input, much easier. The following code snippet demonstrates the different use cases:
The cfg_1 and the cfg_2 will result in the exact same configuration of the alignment where aligning the front of either sequence with gaps is not penalised while the back of both sequences is. The order of the arguments is irrelevant. Specifiers initialised with constant booleans can be mixed with those initialised with bool values. If a specifier for a particular sequence end is not given, it defaults to the specifier initialised with std::false_type.
true and the false case. This adds up to 16 different paths the compiler needs to instantiate.SeqAn also offers predefined seqan3::end_gaps configurations that cover the typical use cases.
| Entity | Meaning |
|---|---|
| free_ends_none | Enables the typical global alignment. |
| free_ends_all | Enables overlap alignment, where the end of one sequence can overlap the end of the other sequence. |
| free_ends_first | Enables semi global alignment, where the second sequence is aligned as an infix of the first sequence. |
| free_ends_second | Enables semi global alignment, where the first sequence is aligned as an infix of the second sequence. |
Adapt the code from above and compute the semi-global alignment where both ends of the first sequence can be aligned to gaps without penalising them.
To accomplish our goal we simply add the align_cfg::aligned_ends option initialised with free_ends_first to the existing configuration.
A scoring scheme can be queried to get the score for substituting two alphabet values using the score member function. SeqAn currently supports currently two scoring schemes: seqan3::nucleotide_scoring_scheme and seqan3::aminoacid_scoring_scheme. You can import them with the following includes:
As the names suggests, you need to use the former when scoring nucleotides and the latter when working with aminoacids. You have already used the seqan3::nucleotide_scoring_scheme in the assignments before. The scoring scheme was default initialised which will result in using the Hamming Distance. The scoring schemes can also be configured by either setting a simple scheme consisting of seqan3::match_score and seqan3::mismatch_score or by providing a custom matrix. The amino acid scoring scheme can additionally be initialised with a predefined substitution matrix that can be accessed via the seqan3::aminoacid_similarity_matrix enumeration class.
Similarly to the scoring scheme, you can use the seqan3::gap_scheme to set the gap penalties used for the alignment computation. The default constructed seqan3::gap_scheme sets the score for a gap to -1 and for a gap opening to 0. Note that the gap open score is added to the gap score when a gap is opened within the alignment computation. Therefore setting the gap open score to 0 disables affine gaps. You can pass a seqan3::gap_score and optionally a seqan3::gap_open_score object to initialise the scheme with custom gap penalties. The penalties can be changed later by using the respective member functions seqan3::gap_scheme::set_linear or seqan3::gap_scheme::set_affine.
To configure the scoring scheme and the gap scheme for the alignment algorithm you need to use the seqan3::align_cfg::scoring and the seqan3::align_cfg::gap configurations. The seqan3::align_cfg::scoring is mandatory - similarly to the seqan3::align_cfg::mode configuration. It would be wrong to assume what the default scoring scheme should be. If you do not provide these configurations, the compilation will fail with a corresponding error message. Not providing the gap scheme is ok. In this case the default initialised gap scheme will be used for the alignment computation.
-2 and gap open costs of -9. Use the BLOSUM62 similarity matrix.Only a few parts of our algorithm need to be adapted. First, we use an amino acid scoring scheme and initialise it with the respective similarity matrix. Second, we initialise the gap scheme to represent the affine gap model as given in the assignment. Et voilà, we have computed a pairwise alignment over aminoacid sequences.
So far we have only computed the score, but obviously in many situations the final alignment is required, e.g. when mapping reads and the user wishes to write the alignment to the final SAM/BAM file. In SeqAn you can simply configure what is going to be computed by the alignment algorithm using the seqan3::align_cfg::result configuration.
Accordingly, the alignment algorithm is configured to use the best implementation to obtain the desired result. The following table shows the different outcomes that can be configured:
| Entity | Available result |
|---|---|
| seqan3::with_score | alignment score |
| seqan3::with_back_coordinate | alignment score; back coordinate |
| seqan3::with_front_coordinate | alignment score; back and front coordinate |
| seqan3::with_alignment | alignment score; back and front coordinate; alignment |
The final result is returned as a seqan3::alignment_result object. This object offers special member functions to access the stored values. If you try to access a value, e.g. the alignment, although you didn't specify with_alignment in the result configuration, a static assertion will be triggered during compilation.
-4 and a simple scoring scheme with mismatch -2 and match 4.
In many situations it is not necessary to compute the entire alignment matrix but only a part of it. This has positive impacts on the performance. To limit the computation space the alignment matrix can be bounded by a band. Thus, only the alignment is computed that fits in this band. Note that this must not be the optimal alignment but in many cases we can give a rough bound on how similar the sequences will be and therefor use the banded alignment. To do so, you can use a seqan3::static_band. It will be initialised with a seqan3::lower_bound and a seqan3::upper_bound. To configure the banded alignment you need to use the seqan3::align_cfg::band configuration.
-3 and upper bound set to 8. How does the result change?
A special form of the pairwise sequence alignment is the edit distance. This distance metric counts the number of edits necessary to transform one sequence into the other. The cost model for the edit distance is fixed. In particular, the match score is 0 and the scores for a mismatch and a gap is -1. Due to the special metric a fast bitvector implementation can be used to compute the edit distance. This happens in SeqAn automatically if the respective configurations are used. To do so, you need a scoring scheme initialised with Manhattan distance (at the moment only seqan3::nucleotide_scoring_scheme supports this) and a gap scheme initialised with -1 for a gap and 0 for a gap open score and computing a seqan3::global_alignment. To make the configuration easier, we added a shortcut called seqan3::align_cfg::edit.
The edit distance can be further refined using seqan3::align_cfg::aligned_ends to also compute a semi-global alignment and the seqan3::align_cfg::max_error configuration to give an upper limit of the allowed number of edits. If the respective alignment could not find a solution within the given error bound, the resulting score is infinity (corresponds to std::numeric_limits::max). Also the alignment and the front and back coordinates can be computed using the align_cfg::result option.
Compute all pairwise alignments from the assignment 1 (only the scores). Only allow at most 7 errors and filter all alignments with 6 or less errors.
Chaining the configurations to build an individual alignment algorithm is a strong advantage of this design. However, some combinations would result in an invalid alignment configuration. To explicitly prevent this we added some security details. First, if a combination is invalid (for example by providing the same configuration more than once) a static assert will inform you about the invalid combination. Here you can find a table depicting the valid configurations. Further, if the seqan3::align_pairwise is called, it checks if the input data can be used with the given configuration. For example, a static assertion is emitted if the alphabet types of the sequences together with the provided scoring scheme do not model seqan3::scoring_scheme. Other possible errors are invalid band settings where the initialised band does not intersect with the actual alignment matrix (the lower diagonal starts beyond the end of the first sequence).
 1.8.16
1.8.16