Class
FragmentStore
Multi container to store contigs, reads, multiple read alignments and genome annotations.
The FragmentStore is a data structure specifically designed for read mapping,
genome assembly or gene annotation. These tasks typically require lots of data structures that
are related to each other like: reads, mate-pairs, reference genome; pairwise alignments; genome annotation.
The FragmentStore subsumes all these data structures in an easy to use interface.
It represents a multiple alignment of millions of reads or mate-pairs against a reference genome consisting
of multiple contigs. Additionally, regions of the reference genome can be annotated with features like 'gene',
'mRNA', 'exon', 'intro' or custom features. The FragmentStore supports I/O functions to read/write a read
alignment in Sam or Amos format and to read/write annotations in Gff/Gtf format.
The FragmentStore can be compared with a database where each table (called "store") is implemented as a
String member of the FragmentStore class. The rows of each table (implemented as structs) are
referred by their ids which are their positions in the string and not stored explicitly.
The only exception is the alignedReadStore whose elements of type AlignedReadStoreElement
contain an id-member as they may be rearranged in arbitrary order, e.g. by increasing genomic positions or by readId.
Many stores have an associated name store to store element names. Each name store is a StringSet that stores the
element name at the position of its id. All stores are present in the FragmentStore and empty if unused. The concrete types,
e.g. the position types or read/contig alphabet, can be easily changed by defining a custom config struct which is a template
parameter of the FragmentStore class.
Include Headers
seqan/store.h
Parameters
The specializing type. Default: | |
The configuration struct. Default: |
Typedefs
| Type of the alignedReadStore member. | |
| Type of the alignedReadTagStore member. | |
| Type of the alignQualityStore member. | |
| Type of the annotationKeyStore member. | |
| Type of the annotationNameStore member. | |
| Type of the annotationStore member. | |
| Type of the annotationTypeStore member. | |
| Type of the contigFileStore member. | |
| Type of the contigNameStore member. | |
| Type of the contigStore member. | |
| Type of the libraryNameStore member. | |
| Type of the libraryStore member. | |
| Type of the matePairNameStore member. | |
| Type of the matePairStore member. | |
| Type of the readNameStore member. | |
| Type of the readSeqStore member. | |
| Type of the readStore member. |
Data Members
| String that stores | |
| StringSet that maps from | |
| String that maps from | |
| StringSet that maps from | |
| StringSet that maps from | |
| String that maps from | |
| StringSet that maps from | |
| String that maps from | |
| StringSet that maps from | |
| String that maps from | |
| StringSet that maps from | |
| String that maps from | |
| StringSet that maps from | |
| String that maps from | |
| StringSet that maps from | |
| StringSet that maps from | |
| String that maps from |
Functions
| Appends an aligned read entry to a fragment store. | |
| Appends two paired-end reads to a fragment store. | |
| Appends a read to a fragment store. | |
| The begin of a container. | |
| Calculates a string with insert sizes for each pair match. | |
| Calculates a string that maps the | |
| Removes all contigs from a fragment store. | |
| Removes all reads from a fragment store. | |
| Removes invalid aligned reads and rename | |
| Renames | |
| Converts all matches to a multiple global alignment in gap-space. | |
| Converts pair-wise alignments to a multiple global alignment. | |
| The end of a container. | |
| Get the "clear" range of a read alignment. | |
| Returns the mate number of read for a given | |
| Returns the read with the given | |
| Calculates a visible layout of aligned reads. | |
| Manually loads a contig sequence. | |
| Loads contigs into fragment store. | |
| Loads reads into fragment store. | |
| Locks a contig sequence from being removed. | |
| Locks all contig sequences from being removed. | |
| Prints a window of the visible layout of reads into a outstream. | |
| Loads records from a file. | |
| Removes a previous contig lock and clears sequence no further lock exist. | |
| Removes a previous lock for all contigs and clears sequences without lock. | |
| Removes a previous contig lock. | |
| Removes a previous lock for all contigs. | |
| Saves records to a file. | |
| Write contigs from fragment store into file. |
Examples
Load read alignments and a reference genome and display the multiple alignment in a genomic range:
File "store_example.cpp"
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 |
The staircase alignment looks as follows:
ATTTAA AATTACAAAATATAGTTGAAAGCTCTAACAATAGA AACCAAGCAGAAGAAAGAGGTTCAGAACTTGAAGA AGTCTCTTATGAATTAA
ATTTA GAAATTACAAAATATAGTTGAAAGCTCTAACAATA ACTAAACCAAGCAGAAGAAAGAGGTTCAGAACTTG AGACAAGTCTCTTATGAATTAA
attta GAAATTACAAAATATAGTTGAAAGCTCTAACAATAG AACCAAGCAGAAGAAAGAGGCTCAGAACTTGAAGA AGTCTCTTATGAATTAA
ATTTAA ATTACAAAATATAGTTGAAAGATCTAACAATAGAC CCAAGCAGAAGAAAGAGGTTCAGAACTTGAAGACAA TTATGAATTAA
ATTTAAGAA TTACAAAATATAGTTGAAAGCTCTAACAATAGACT AAGCAGAAGAAAGAGGTTCAGAACTTGAAGACAAG TATGAATTAA
ATTTAAGAAA ACAAAATATAGTTGAAAGCTCTAACAATAGACTAA GCAGAAGAAAGAGGTTCAGAACTTGAAGACAAGTC ATGAATTAA
ATTTAAGAAA ACAAAATATAGTTGAAAGCTCTAACAATAGACTAA CAGAAGAAAGAGGTTCAGAACTTGAAGACAAGTCT TGAATTAA
ATTTAAGAAA ACAAAATATAGTTGAAAGCTCTAACAATAGACTAA CAGAAGAAAGAGGTTCANANNNTGANGACAAGTCT TGAATTAA
ATTTAAGAAATT CAAAATATAGTTGAAAGCTCTAACAATAGACTAAA GAAGAAAGAGGTTCAGAACTTGAAGACAAGTCTCT GAATTAA
ATTTAAGAAAT AAAATATAGTTGAAAGCTCTAACAATAGACTAAAC AAGAAAGAGGTTCAGAACTTGAAGACAAGTCTCGT GAATTAA
ATTTAAGAAAT AAAATATAGTTGAAAGCTCTAACAATAGACTAAAC AAGAAAGAGGTTCAGAACTTGAAGACAAGTCTCTT AATTAA
ATTTAAGAAAT AAATATAGTTGAAAGCTCTAACAATAGACTAAACC GAAAGAGGTTCAGAACTTGAAGACAAGTCTCTTATG
ATTTAAGAAATT AAATATAGTTGAAAGCTCTAACAATAGACTAAACC AAGAGGTTCAGAACTTGAAGACAAGTCTCTTATGA
ATTTAAGAAATT AATATAGTTGAAAGCTCTAACAATAGACTAAACCAA AAGAGGTTCAGAACTTGAAGACAAGTCTCTTATGA
ATTTAAGAAATTACA ATATAGTTGAAAGCTCTAACAATAGACTAAACCAA GAGGTTCAGAACTTGAAGACAAGTCTCTTATGAAT
ATTTAAGAAATTACAA ATAGTTGAAAGCTCTAACAATAGACTAAACCAAGC GAGGTTCAGAACTTGAAGACAAGTCTCTTATGAAT
ATTTAAGAAATTACAAAATA AGTTGAAAGCTCTAACAATAGACTAAACCAAGCAG AGGTTCAGAACTTGAAGACAAGTCTCTTATGAATT
ATTTAAGAAATTACAAAATAT TTGAAAGCTCTAACAATAGACTAAACCAAGCAGAA GGTTCAGAACTTGAAGACAAGTCTCTTATGAATTA
ATTTAAGAAATTACAAAATATA GAAAGCTCTAACAATAGACTAAACCAAGCAGAAGAAAGAG TTCAGAACTTGAAGACAAGTCTCTTATGAATTAA
ATTTAAGAAATTACAAAATATAGTTGAA CTAACAATAGACTAAACCAAGCAGAAGAAAGAGTT CTTGAAGACAAGTCTCTTATGAATTAA
ATTTAAGAAATTACAAAATATAGTTGAAA CTAACAATAGACTAAACCAAGCAGAAGAAAGAGGTT TTGAAGACAAGTCTCTTATGAATTAA
ATTTAAGAAATTACAAAATATAGTTGAAAG TAACAATAGACTAAACCAAGCAGAAGAAAGAGGTT TGAAGACAAGTCTCTTATGAATTAA
ATTTAAGAAATTACAAAATATAGTTGAAAGCTCT ACAATAGACTAAACCAAGCAGAAGAAAGAGGTTCA TGAAGACAAGTCTCTTATGAATTAA
TTAAGAAATTACAAAATATAGTTGAAAGCTCTAAC GACTAAACCAAGCAGAAGAAAGAGGTTCAGAACTT AAGACAAGTCTCTTATGAATTAA
TAAGAAATTACAAAATATAGTTGAAAGCTCTAACAATAGA GGTTCAGAACTTGAAGACAAGTCTCTTATGAATTA
TTACAAAATATAGTTGAAAGCTCTAACAATAGACT GGTTCAGAACTTGAAGACAAGTCTCTTATGAATTA
ATAGTTGAAAGCTCTAACAATAGACTAAACCAAGC GTTCAGAACTTGAAGACAAGTCTCTTATGAATTAA
AAAGCTCTAACAATAGACTAAACCAAGCAGAAGAA TCAGAACTTGAAGACAAGTCTCTTATGAATTAA
AAAGCTCTAACAATAGACTAAACCAAGCAGAAGAA NAAGACAAGTCTCTTATGAATTAA
AAGCTCTAACAATAGACTAAACCAAGCAGAAGAAA GAAGACAAGTCTCTTATGAATTAA
TAACAATAGACTAAACCAAGCAGAAGAAAGAGGTT AGTCTCTTATGAATTAA
TAACAATAGACTAAACCAAGCAGAAGAAAGAGGTT GTCTCTTATGAATTAA
AACAATAGACTAAACCAAGCAGAAGAAAGAGGTTC
AACAATAGACTAAACCAAGCAGAAGAAAGAGGTTC
AATAGACTAAACCAAGCAGAAGAAAGAGGTTCAGA
AATAGACTAAACCAAGCAGAAGAAAGAGGTTCAGA
The following figures visualize the relations between the different stores:
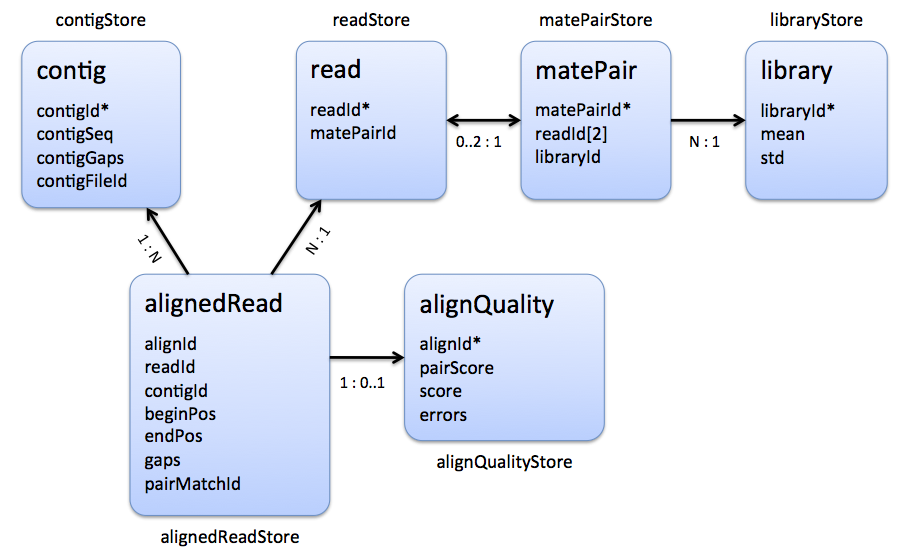 |
| Stores that are involved in the representation of a multiple read alignment. |
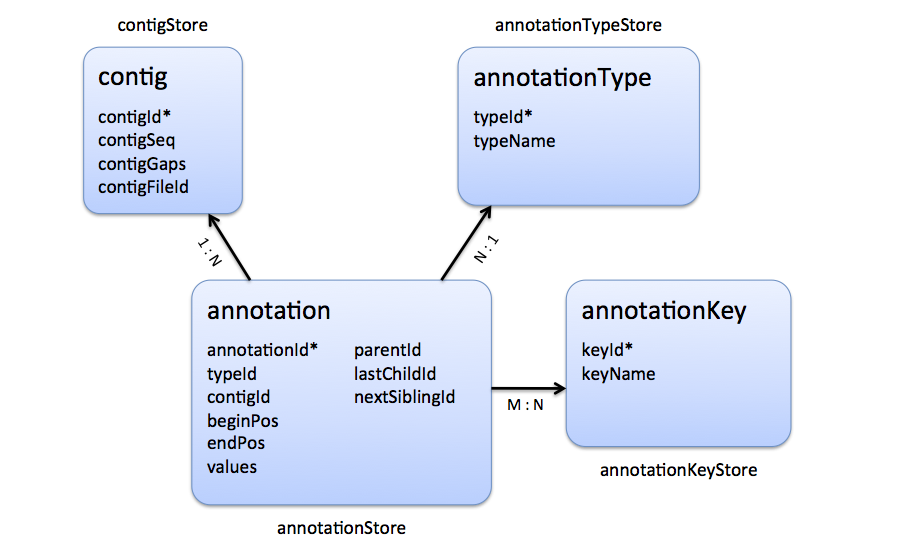 |
| Stores that are involved in the representation of a genome alignment. |
SeqAn - Sequence Analysis Library - www.seqan.de